System blowing up - and running with polymers/peptides #41
Replies: 2 comments 19 replies
-
|
Did you run prepare_dual_topologies.py with the tutorial inputs? Your system is likely blowing up during the first minimization, which is odd. Would you mind checking that the system is sane (eg, by taking a look at it in pymol)? |
Beta Was this translation helpful? Give feedback.
-
|
I ran again from scratch with 2021 version of gromacs (MPI enabled) using the tutorial files it worked fine, but in the case of 2022 version of gromacs I don't know why I am still getting the same error again (haven't tried my own molecules yet) . |
Beta Was this translation helpful? Give feedback.
-
|
I wouldn't expect that version 2022 to behave in a different way than 2021 for the same system. But I am glad to know that it worked. I wrote PyAutoFEP mainly for small organic molecules, but there is no hard limit that prevents it to work with larger molecules. I guess that the most relevant problem for peptides or nucleic acids is that you would need to use the macromolecule FF to describe the ligand, while PyAutoFEP expects a ligand topology as input. Here's an idea: use a toll like parmed to convert/export the peptide/DNA/RNA topology a single file. Then use it as lig topology input to The MCS algorithm may take a long time for large molecules and you may want to use the 3d MCS (but the default, graph-based MCS may work as well). Passing the MCS as smiles can greatly decrease the PS: there is a long-term goal to add a decent support for peptides in PyAutoFEP. It will use some of the code that I am writing for the charged perturbations, so peptide-specific code is not seeing the light of the day any time soon. In case you wanna give the regular code a try using peptides, feel free to write me here or by email. I would love to help you (and test the code for this use case in the process). |
Beta Was this translation helpful? Give feedback.
-
|
Thank you so much for your advice. |
Beta Was this translation helpful? Give feedback.
-
|
Sure thing. It's [email protected]. Feel free to write me. I guess polymers would actually be easier than peptides/DNA/RNA. On the other hand, supplying an MCS will very likely be required (see https://www.rdkit.org/docs/GettingStartedInPython.html#maximum-common-substructure). |
Beta Was this translation helpful? Give feedback.
-
|
Thank you for the suggestion I will look into MCS while preparing the systems. |
Beta Was this translation helpful? Give feedback.
-
|
Hello, I'm having a new problem where all of the simulations are running fine, but the 'rerun' step is not running for some reason. |
Beta Was this translation helpful? Give feedback.
-
|
Thank you for the suggestions, I ll look into these and come back to you. |
Beta Was this translation helpful? Give feedback.
-
|
I analyzed the files and the error is in the FullSystem.pdb, the dual molecule is not correct. |
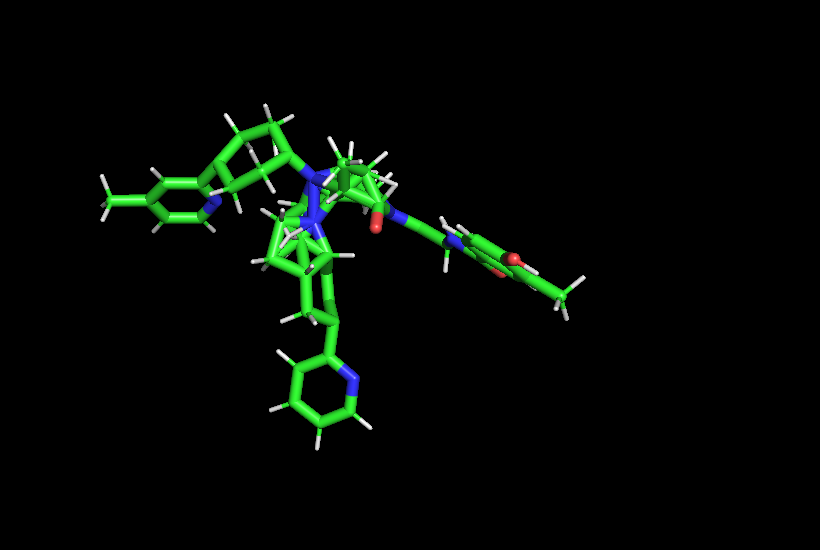
Beta Was this translation helpful? Give feedback.
-
|
From this image it is not clear whether the dual-molecule is correct or not. Because in the dual-topology module, atoms from both A and B states are modelled, pymol will render all atoms, leading to incorrect structures. One way to assess the dual-topology is from the MCS data produced by Anyway, from the structure you seem to be perturbing, you seem to be trying to perturb a quite large structure to smaller one. Perturbing this many atoms will likely require more than 12 lambda windows and maybe a longer equilibration. Also, make sure that the binding does allow this large perturbation. |
Beta Was this translation helpful? Give feedback.
-
|
So, I was trying with different combinations for my system in which got again a segment fault when I see the structure the atoms of the ligands is floating in the system, I have attached the image, I am not sure what is happening. |
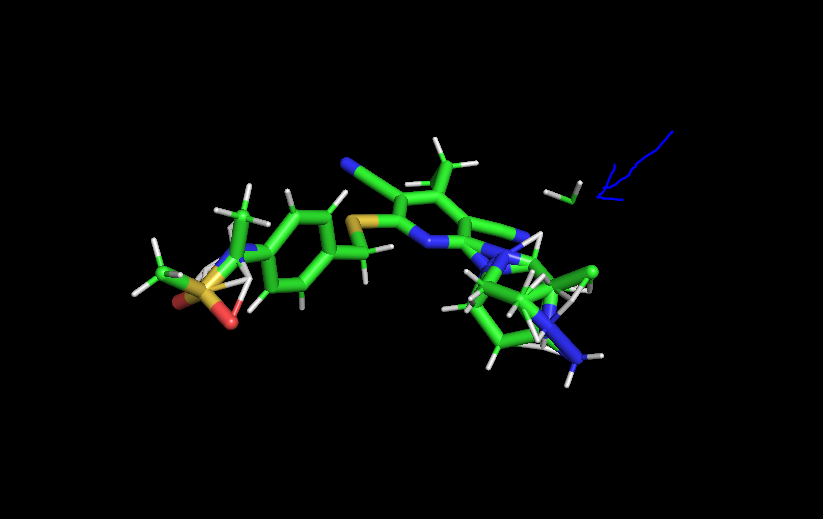

Beta Was this translation helpful? Give feedback.
-
|
The disconnected CH2 maybe a artifact of the PyMOL representation (it draws bonds based on atomic distance) or maybe a problem in the dual-topology generated by PyAutoFEP. Is the second image a representation of the system after the crash or the input structure? If it's the latter, that's almost certainly a bug in Would you mind extracting the A and B molecules so we can better inspect what's going on? Or sharing the starting molecules and the full output of the If you'd rather get a closer look yourself, look for the for the MCS info in |
Beta Was this translation helpful? Give feedback.
Uh oh!
There was an error while loading. Please reload this page.
-
Hi, I was following the tutorial that is provided while running the final bash script for the protein part I, am getting this error.
ERROR 1 [file FXR_12-FXR_74/protein/lambda0/min01/min01.mdp]:
The largest distance between excluded atoms is 1.063 nm, which is larger
than the cut-off distance. This will lead to missing long-range
corrections in the forces and energies.
any comments on the same that why I am getting this error will be beneficial.
Beta Was this translation helpful? Give feedback.
All reactions