{kind=link}
Installation | Enviroment | Example | Advanced Features | Input | Output | Parameter | Citation | Contact
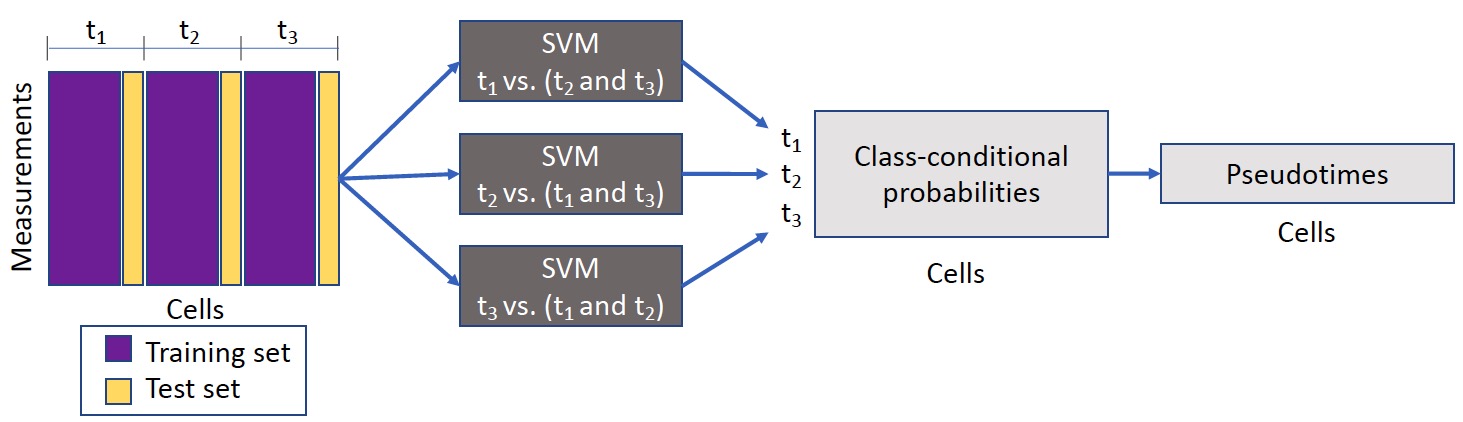
Sceptic can perform pseudotime analysis on various types of single-cell/single-nucleus data. The model takes as input a collection of single-cell/single-nucleus data and then learns the relationship between the observed data and the associated time stamps, and finally uses the trained model to assign to each cell a real-valued pseudotime. Ideally, the pseudotimes assigned by Sceptic reflect each cell's progression along a notion of time---developmental, cell cycle, disease progression, aging---that is appropriate to the given data. Ideally, the pseudotimes assigned by Sceptic reflect each cell's progression along a notion of time---developmental, cell cycle, disease progression, aging---that is appropriate to the given data.
Sceptic software is available on the Python package index (PyPI), latest version 0.3.3. To install it using pip, simply type:
$ pip install scepticSceptic is associated with the following packages.
- python >= 3.7.7
- numpy >= 1.19.5
- pandas >= 1.3.5
- sklearn >= 1.0.2
We downloaded the processed scGEM dataset from UnionCom’s GitHub page.
$ python test/scGEM/scGEM.pyThe script will generate 4 outputs from Sceptic described in the section above and save it at: test/scGEM/.
Sceptic now automatically handles time label encoding! You can pass actual biological time values directly:
from sceptic import run_sceptic_and_evaluate
# Option 1: Pass actual time values directly (easiest!)
time_labels = np.array([0, 0, 8, 8, 16, 16, 24, 24]) # hours
cm, pred, pseudotime, prob = run_sceptic_and_evaluate(
data, time_labels, method="xgboost"
)
# Option 2: Use encoded labels with time mapping
encoded_labels = np.array([0, 0, 1, 1, 2, 2, 3, 3])
actual_timepoints = np.array([0, 8, 16, 24]) # hours
cm, pred, pseudotime, prob = run_sceptic_and_evaluate(
data, encoded_labels, label_list=actual_timepoints, method="xgboost"
)Benefits:
- ✅ No manual label encoding required
- ✅ Pseudotime values in meaningful biological units
- ✅ More intuitive and less error-prone
- ✅ Backward compatible with existing code
Sceptic also includes utility modules for comprehensive evaluation and publication-quality visualization!
The sceptic.evaluation module provides comprehensive metrics for assessing pseudotime predictions:
from sceptic import evaluation
# Comprehensive evaluation with all metrics
results = evaluation.evaluate_sceptic_results(
confusion_matrix=cm,
y_true=label,
y_pred=label_predicted,
pseudotime=pseudotime,
true_time=true_time_values,
include_regression=True, # Optional: include MAE/MSE
verbose=True
)Available metrics:
- Classification: Accuracy, balanced accuracy, per-class precision/recall
- Correlation: Spearman, Pearson, and Kendall correlations
- Regression (optional): MAE, MSE, RMSE
The sceptic.plotting module provides publication-quality visualizations:
from sceptic import plotting
# Set publication style
plotting.set_publication_style()
# Create confusion matrix heatmap
fig = plotting.plot_confusion_matrix(
confusion_matrix=cm,
label_list=label_list,
output_path="confusion_matrix.png",
dpi=300
)
# Create violin plot
fig = plotting.plot_pseudotime_violin(
pseudotime=pseudotime,
true_labels=time_labels,
output_path="violin_plot.png",
dpi=300
)
# Stratified analysis by groups (e.g., cell types)
plotting.plot_pseudotime_by_group(
pseudotime=pseudotime,
true_labels=time_labels,
group_labels=cell_types,
output_dir="violin_by_cell_type"
)For detailed tutorials, see the examples/ directory:
- basic_usage.ipynb: Introduction to Sceptic workflow
- custom_evaluation.ipynb: Advanced evaluation and visualization
The list of parameters is given below:
eFold: # of folds for external cross-validation (default=3).iFold: # of folds for internal cross-validation (default=4).
For SVM implementation:
kernel: The kernel function for sceptic SVM classfier (default=('linear', 'rbf')). Sklearn supports four kinds of kernels: linear, polynomial, rbf, sigmoid.C: The C parameter for rbf kernel (default=[0.1, 1, 10]). The C parameter trades off correct classification of training examples against maximization of the decision function’s margin. See more details here.
For XGboost implementation:
max_depth: Maximum depth of a tree (default=[3, 5]). Increasing this value will make the model more complex and more likely to overfit. 0 indicates no limit on depth. Beware that XGBoost aggressively consumes memory when training a deep tree. See more details here.learning_rate: Step size shrinkage used in update to prevent overfitting (default=[0.1, 0.3]). After each boosting step, we can directly get the weights of new features, and eta shrinks the feature weights to make the boosting process more conservative. See more details here.
In case the user is providing the input data:
data_concat: the input cell by measurement matrix. (# of cells by # of measurements)label: processed cell time label. (# of cells by 1)label_list: unique list of possible cell time labels. (# of time points by 1)parameters: Sceptic parameter dictionary. (SVM default={'kernel': ('linear', 'rbf'), 'C': [0.1, 1, 10]}; XGboost default= {"max_depth": [3, 5], "learning_rate": [0.1, 0.3], "n_estimators": [100], "subsample": [0.8]})method: "svm" or "xgboost" implementation. For large dataset, we recommend "xgboost" implementation.use_gpu: Only applies if method="xgboost".
When one uses sceptic.run_sceptic_and_evaluate function, several outputs are generated:
cm: the confusion matrix for Sceptic's nested cross-validation. (# of time_points by # of time points)label_predicted: Sceptic's predicted discrete label for each cell. (# of cells by 1)pseudotime: Sceptic's predicted pseudotime (continuous) for each cell. (# of cells by 1)sceptic_prob: the class-proabilities for each cell. (# of cells by # of time points)
In case you have questions, reach out to [email protected].
Pseudotime analysis for time-series single-cell sequencing and imaging data
If you have found our work useful, please consider citing us:
Li, G., Kim, HJ., Pendyala, S. et al. Sceptic: pseudotime analysis for time-series single-cell sequencing and imaging data. Genome Biol 26, 209 (2025). https://doi.org/10.1186/s13059-025-03679-3
@article{li2025sceptic,
title={Sceptic: pseudotime analysis for time-series single-cell sequencing and imaging data},
author={Li, Gang and Kim, Hyeon-Jin and Pendyala, Sriram and Zhang, Ran and Vert, Jean-Philippe and Disteche, Christine M and Deng, Xinxian and Fowler, Douglas M and Noble, William Stafford},
journal={Genome Biology},
volume={26},
pages={209},
year={2025}
}