{kind=link}
{kind=link}
![]()
Molden2AIM is a utility program which can be used to create AIM-WFN, AIM-WFX, and NBO-47 files from a Molden file.
Version 5.1.0 (08/29/2023).
- The MOLDEN file generated by Bagel, CP2k, or eT (since Ver. 1.4) has been supported.
- The
[Nval]data block (suggested by Multiwfn) may be used for ECP basis sets. - Ghost atom in the
[Atoms]block has been supported.
Version 5.0.8 (07/01/2023).
- Bug fix: xenon was identified as a dummy atom by mistake.
Version 5.0.7 (04/23/2023).
- A new option
ANSIfor colors in terminal. - For the Molden file generated by Molpro, energy is printed in the WFN and WFX files.
- For the Molden file generated by BDF, energy and virial ratio are printed in the WFN and WFX files.
- Bug fix for the option
ALLMO: abs(occ) is checked now, which is important for natural orbitals.
Version 5.0.6 (11/12/2021).
- Bug fix for reading MO coefficients printed in scientific notation.
Version 5.0.5 (07/23/2021).
- In the title section of new-MOLDEN/WFN/WFX/47 files, print the hostname and the original MOLDEN file name with the full path by setting title=1 in m2a.ini.
- Bug fix. A space between index and coefficient in the
[MO]data block may be missing in some MOLDEN files, which is completed. - Bug fix. Negative
nosuppinm2a.iniwas omitted by mistake. - A command line parameter
-ihas been added.
Version 5.0.4 (02/07/2021).
- Bug fix. The
[CORE]/[PSEUDO]data block was omitted by mistake in subroutine ROADrv. - Bug fix. In the new MOLDEN file, ZA instead of ZA-Ncore is printed now in the
[ATOMS]data block.
Version 5.0.3 (01/30/2021).
- Improved compatibility with GNU gfortran 10.
Version 5.0.2 (10/09/2020).
- The MOLDEN file with H-functions has been supported if it is generated by Dalton.
- The utility
ReOrdAtmhas been merged into Molden2AIM and runs automatically. - For the MOLDEN file saved by ORCA,
[PROGRAM] orcain MOLDEN andprogram=1inm2a.iniare not needed in the case of the default titleMolden file created by orca_2mkl for BaseName=....
Version 5.0.0 (06/05/2020).
- If possible, save a new MOLDEN file or NBO-47 file in spherical functions.
carsph=1inm2a.iniis required. - The MOLDEN file with H-functions has been supported if it is generated by Multiwfn, ORCA, or CFour (Ver. 2.1).
- If possible, the
$LCAOMOand$FOCKblocks will be printed in the NBO-47 file (nbopro=1inm2a.iniis required), so the Second Order Perturbation Theory Analysis may be performed by NBO for the RHF, UHF, RKS, and UKS types of wavefunctions. - Orthogonality will be checked if the
$FOCKblock exists in the NBO-47 file. - Bug fix for modern Fortran compilers.
Version 4.4.0 (05/27/2020).
- A new X2C/PBE0 EDF library (by Chun Gao) can take core correlations into account, which may be requested by
edftyp=1inm2a.ini. Some test calculations of noble gas atoms with 22 functionals showed that PBE0 can reproduce the core densities of CCSD(T,full) with the best agreements. - The initialization file
m2a.inimay be generated automatically if it doesn't exist. - The fitting program denfit.f90 has been modified to improve the accuracy.
- Bug fix: energies in the WFN file were wrong.
Version 4.3.0 (02/09/2019).
- The Molden file generated by StoBe has been supported.
- The Molden file generated by Crystal (molecule only) has been supported through
[Program] crystalin MOLDEN orPROGRAM=10in m2a.ini. - The number of core electrons may also be specified in the terminal.
Version 4.2.1 (05/11/2018).
- The EDF library has been updated for the following cores/elements: ncore = 2 (B), 10 (Na), 28 (Cu, Pd, I, Xe, Cs, Sm, Eu, Gd, Tb), 46 (Cd, Xe), 78 (Pa, Es, Fm), and 92 (Cn, Nh). It's found that these old EDFs may produce a local minimum at R = 0 and lead to a (3,+3) critical point wrongly. Thank Dr. Tian Lu for reporting the problem.
- The fitting program denfit.f90 has been modified for the above problem.
- It converts the data format from Molden to AIM's WFN. The latter format can be read by AIMPAC, AIMPAC2, AIM2000, AIMALL, AIM-UC, Critic2, DensToolKit, DGrid, MORPHY98, Multiwfn, ORBKIT, PAMoC, ProMolden, TopChem, TopMoD, Xaim, and so on. The GAB file of Gabedit is compatible.
- It saves NBO's *.47 data file. One can do NBO analysis using the stand-alone GENNBO program. In addition, the following loops can be performed using NBO or NBO2Molden. However the results may be different since NBO saves natural bond orbitals (NBOs) into the MOLDEN file by default.
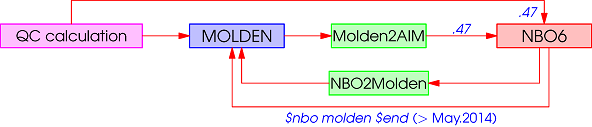
- After the *.47 file being generated, it can calculate the generalized Wiberg bond order indices (GWBO) in MO (see I. Mayer, C.P.L. 97, 270, 1983). In the case of closed-shell system, they are the Mayer bond orders (MBO) in MO.
- It saves AIM's WFX data file, which can be read by AIMALL, Critic2, DensToolKit, GPView, Multiwfn, or ORBKIT. There are two versions of atomic EDF library for Z = 3-120 controlled by
edftypinm2a.ini: the default X2C/HF version byedftyp=0(see W. Zou, Z. Cai, J. Wang, K. Xin, An open library of relativistic core electron density function for the QTAIM analysis with pseudopotentials, J. Comput. Chem. 2018, 39, 1697-1706) and the X2C/PBE0 version byedftyp=1.
> F90 -O3 edflib.f90 edflib-pbe0.f90 molden2aim.f90 -o molden2aim.exe
where F90 can be gfortran, nvf90 (pgf90), ifort, or other Fortran90 compilers.
- Windows
- Put
molden2aim.exeand MOLDEN/Gabedit files into the same folder. - If necessary, insert a
[Program] program_nameline into the MOLDEN file, or edit theprogramparameter inm2a.ini(you can also setup other parameters there). - If ECP or MCP is used, insert a
[Core]or[Pseudo]segment into the MOLDEN/Gabedit file. See below for the format and examples. - Double-click
molden2aim.exe, and then type in the MOLDEN/Gabedit file name.
- Unix/Linux/MacOS
-
Put
molden2aim.exeand MOLDEN/Gabedit files into the same folder. -
If necessary, insert a
[Program] program_nameline into the MOLDEN file, or edit theprogramparameter inm2a.ini(you can also setup other parameters there). -
If ECP or MCP is used, insert a
[Core]or[Pseudo]segment into the MOLDEN/Gabedit file. See below for the format and examples. -
In the terminal, type in
./molden2aim.exe
and then type in the MOLDEN/Gabedit file name, or provide the MOLDEN/Gabedit file name in command line
./molden2aim.exe -i MOLDEN_FILE_NAME
In the case of ECP or MCP, a data block of [Core] should be defined in the MOLDEN file. The format is
[Core]
Iatom : Ncore or Element: Ncore
...
where Ncore is the number of core electrons replaced by ECP or MCP. Atom/element with Ncore=0 can be ignored. For example, for a cluster with the atoms N_1, N_2, N_3, Pt_4, and Pt_5, it can be
[Core]
Pt: 60
N : 2
2 : 0
This means that the numbers of core electron are 60 in Pt_4 and Pt_5 and 2 in N_1 and N_3. In N_2 the number of core electron is set to 2 but then reset to 0. It is equivalent to
[Core]
1 : 2
3 : 2
4 : 60
5 : 60
Another way is to define a data block of [Pseudo] in the MOLDEN file, which is supported by Molden. The format is
[Pseudo]
Name1 IAtom1 ZA1-Ncore1
Name2 IAtom2 ZA2-Ncore2
...
Starting from Version 5.1.0, the [Nval] block suggested by Multiwfn may also be used.
[Nval]
Element1 nval1 (nval = ZA - Ncore)
Element2 nval2
...
Ghost atoms in the MOLDEN file may be specified by a prefix bq-, a prefix ghost-, a suffix -bq, a suffix -ghost (case insensitive), or atomic_number = 0. In the following example, all the five carbon atoms are ghost ones.
[Atoms] AU
C 1 0 0.0000000 2.6361503 0.0000000
C-bq 2 6 -2.2829731 1.3180752 0.0000000
C-ghost 3 6 -2.2829731 -1.3180752 0.0000000
bq-C 4 6 0.0000000 -2.6361503 0.0000000
ghost-C 5 6 2.2829731 -1.3180752 0.0000000
...
MOLDEN (or GAB) files generated by the the following programs are fully or partly supported by Molden2AIM at present.
- ACES-II, (> 2.9)
- Bagel
- BDF-G
- CADPAC
- CFour
- Columbus
- CP2k, (molecule using GTFs only)
- Crystal, (molecule only)
- DALTON, (> 2013)
- deMon2k
- eT, (>= Ver. 1.4)
- Firefly, through the utility Molden or Gabedit. See molden_gabedit.jpg.
- Gaussian, through the utility Molden or Gabedit. See molden_gabedit.jpg.
- Gamess, through the utility Molden or Gabedit. See molden_gabedit.jpg.
- Gamess-UK, through the utility Molden. See molden_gabedit.jpg.
- Jaguar
- MOLCAS
- MOLPRO
- MRCC
- Multiwfn
- NBO, (> May.2014)
- NWChem, (>= Ver. 6.8) by MOLDEN_NORM JANPA or NONE to generate a MOLDEN file. See the attached examples.
- ORCA
- Priroda
- PSI4
- PySCF
- Q-Chem
- StoBe
- TeraChem
- Turbomole
{kind=link}
See readme.html for details.
Examples of applications can be found in W. Zou, D. Nori-Shargh, and J. E. Boggs, On the Covalent Character of Rare Gas Bonding Interactions: A New Kind of Weak Interaction, J. Phys. Chem. A 117, 207-212 (2013); Erratum: J. Phys. Chem. A 120, 2057-2057 (2016).
The EDF library (X2C/HF version) was published in W. Zou, Z. Cai, J. Wang, and K. Xin, An open library of relativistic core electron density function for the QTAIM analysis with pseudopotentials, J. Comput. Chem. 39, 1697-1706 (2018).