scRNAtoolVis documentation.
This package mainly is used to replot seurat default plot and with other interesting functions. I will add other functions into this package in the future.
clusterCornerAxes is used to add corner axis on the left-bottom UMAP/tSNE principle component plot.
We load test data in scRNAtoolVis package:
test <- system.file("extdata", "seuratTest.RDS", package = "scRNAtoolVis")
tmp <- readRDS(test)default plot:
# umap
clusterCornerAxes(object = tmp,reduction = 'umap',
noSplit = T)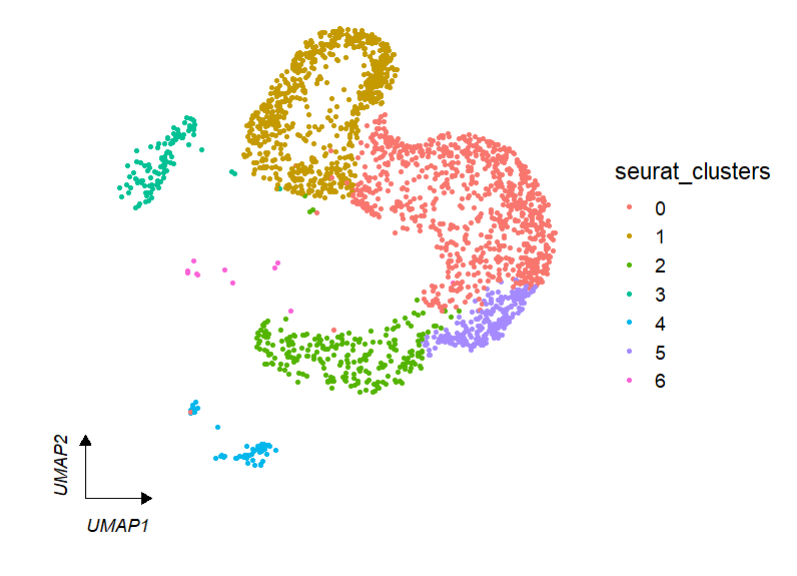
We can change arrow type:
# arrowType
clusterCornerAxes(object = tmp,reduction = 'umap',
noSplit = T,arrowType = 'open')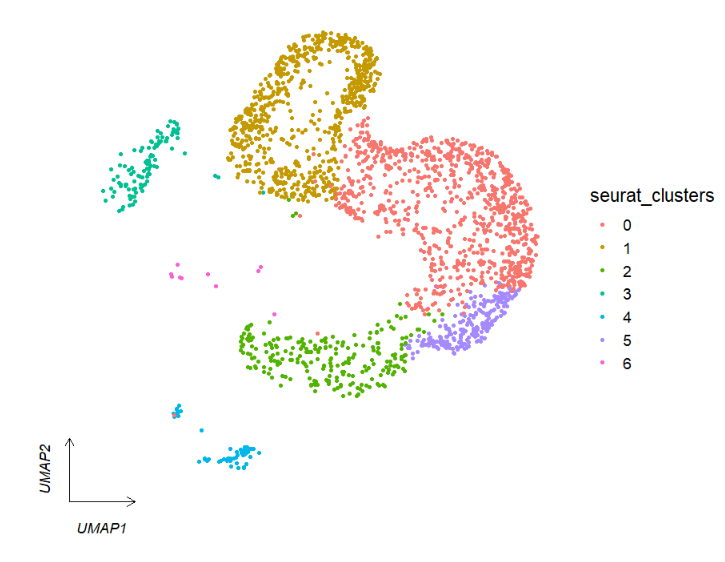
We can facet by seurat metadata column catogary variable:
# facet by metadata column "orig.ident"
clusterCornerAxes(object = tmp,reduction = 'umap',
noSplit = F,groupFacet = 'orig.ident',
relLength = 0.5)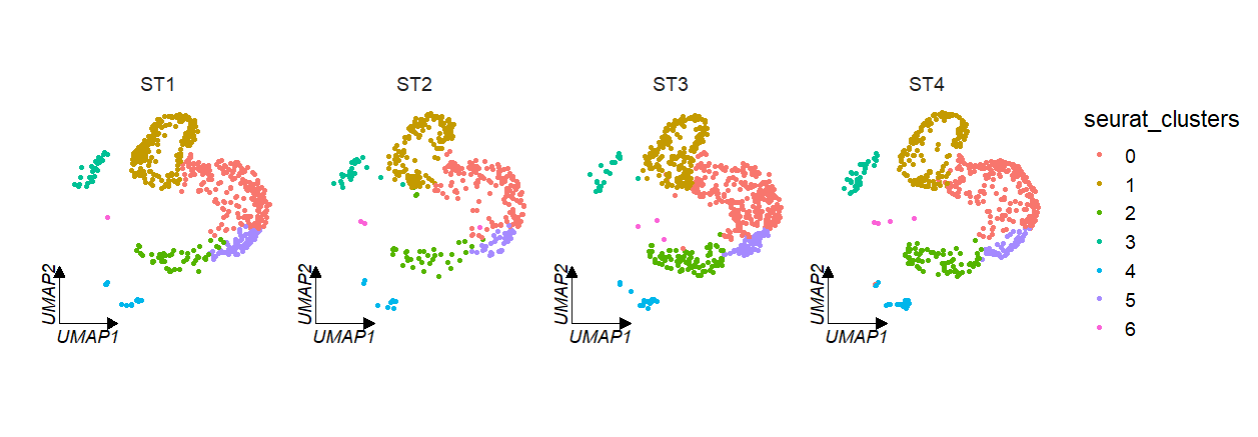
If multiple corner axises will confuse you, you can also set axes = 'one' to retain only one axis on the left:
# retain only one axes
clusterCornerAxes(object = tmp,reduction = 'umap',
noSplit = F,groupFacet = 'orig.ident',
relLength = 0.5,
axes = 'one')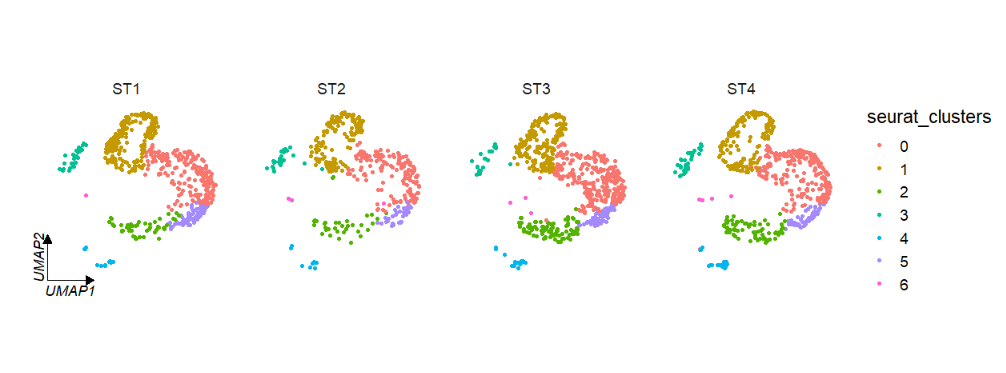
Change the axis and label color:
# line color
clusterCornerAxes(object = tmp,reduction = 'umap',
noSplit = F,groupFacet = 'orig.ident',
relLength = 0.5,
lineTextcol = 'grey50')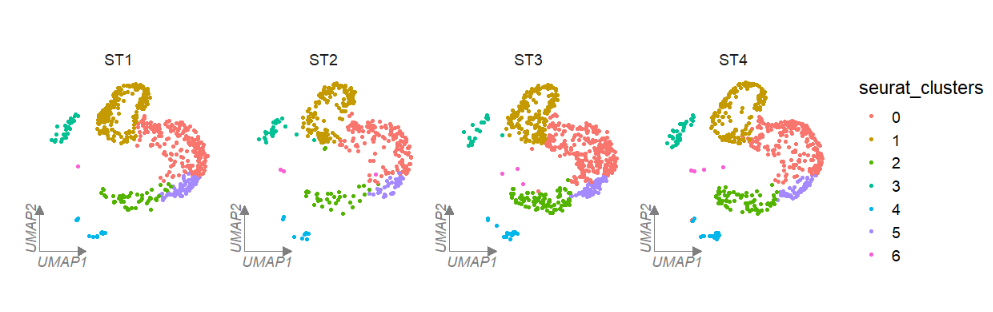
Use tSNE reduction data:
# tsne
clusterCornerAxes(object = tmp,reduction = 'tsne',
noSplit = F,groupFacet = 'orig.ident',
relLength = 0.5)
Change a cool theme:
clusterCornerAxes(object = tmp,
reduction = 'umap',
noSplit = F,
groupFacet = 'orig.ident',
relLength = 0.5,
cornerTextSize = 4,
themebg = 'bwCorner')
FeatureCornerAxes is used to add corner axises on the gene expression reduction map:
# umap
FeatureCornerAxes(object = tmp,reduction = 'umap',
groupFacet = 'orig.ident',
relLength = 0.5,relDist = 0.2,
features = c("Actb","Ythdc1", "Ythdf2"))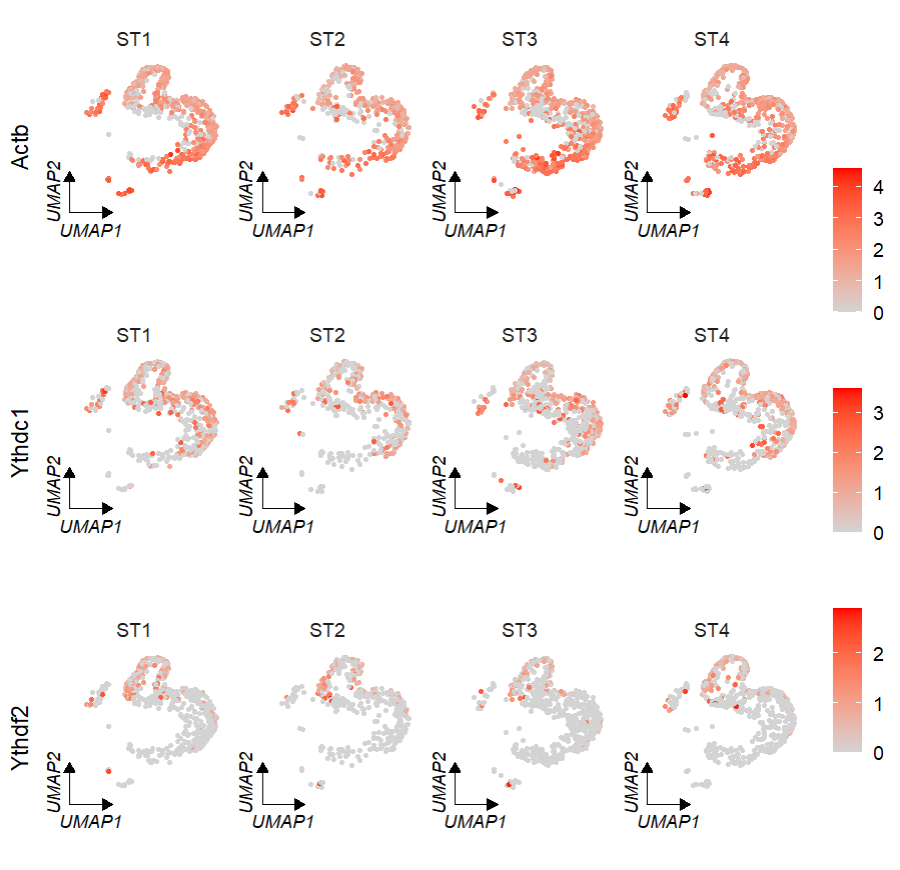
Keep one axis:
# one axes
FeatureCornerAxes(object = tmp,reduction = 'umap',
groupFacet = 'orig.ident',
features = c("Actb","Ythdc1", "Ythdf2"),
relLength = 0.5,relDist = 0.2,
axes = 'one',
lineTextcol = 'grey50')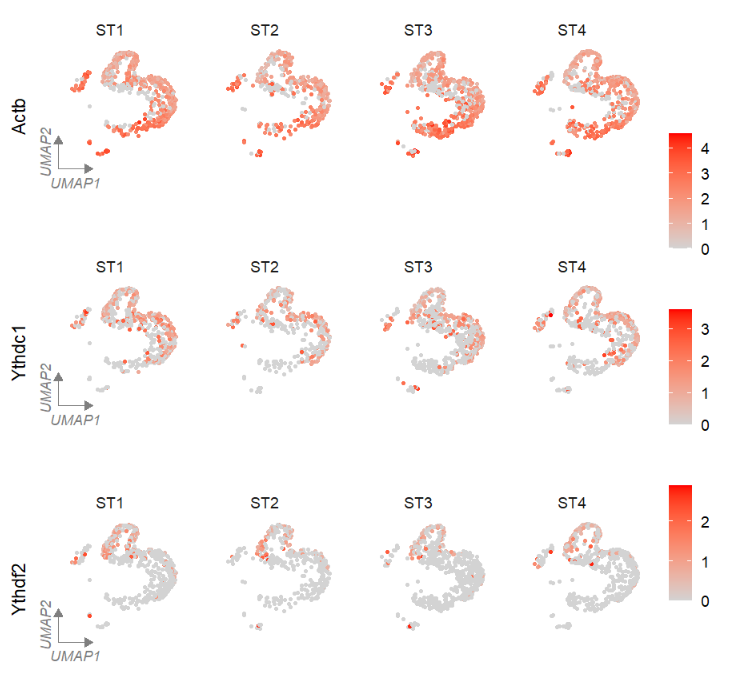
tSNE reduction:
# tsne
FeatureCornerAxes(object = tmp,reduction = 'tsne',
groupFacet = 'orig.ident',
relLength = 0.5,relDist = 0.2,
features = c("Actb","Ythdc1", "Ythdf2"))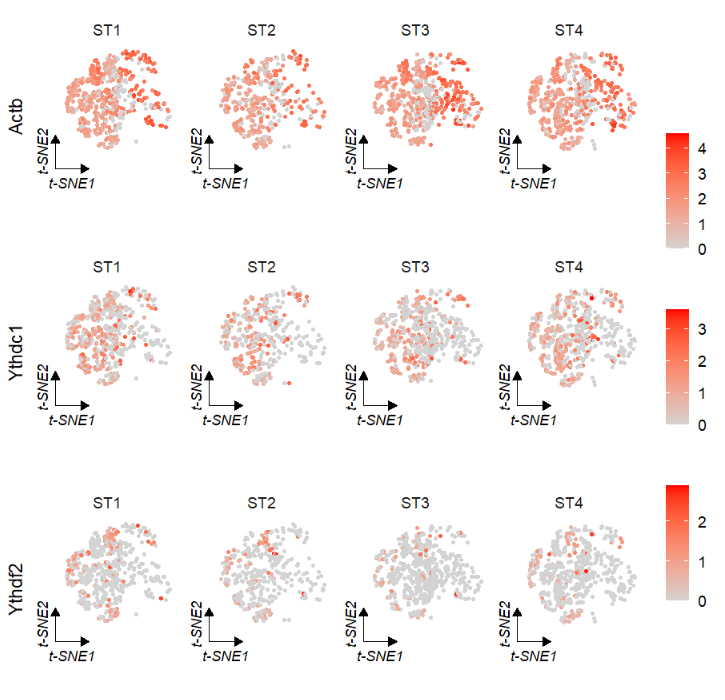
Change a cool theme:
# umap
FeatureCornerAxes(object = tmp,reduction = 'umap',
groupFacet = 'orig.ident',
features = c("Actb","Ythdc1"),
cornerTextSize = 3,
themebg = 'bwCorner')
Add circle on clusters:
# add circle
clusterCornerAxes(object = tmp,reduction = 'umap',
noSplit = T,
cornerTextSize = 3.5,
themebg = 'bwCorner',
addCircle = TRUE,
cicAlpha = 0.2,
nbin = 200)
Relative parameters to control the circle as follows:

AverageHeatmap is used to plot averaged expression cross cluster cells.
load data:
httest <- system.file("extdata", "htdata.RDS", package = "scRNAtoolVis")
pbmc <- readRDS(httest)
# load markergene
markergene <- system.file("extdata", "top5pbmc.markers.csv", package = "scRNAtoolVis")
markers <- read.table(markergene, sep = ',', header = TRUE)plot:
# plot
AverageHeatmap(object = pbmc,
markerGene = markers$gene)
change color:
# change color
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
htCol = c("#339933", "#FFCC00", "#FF0033"))
Supporting with your own cluster colors by annoCol = TRUE and myanCol:
# change annotation color
library("scales")
library(ggsci)
mycol <- hue_pal()(9)
mycol1 <- pal_npg()(9)
# plot
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
annoCol = TRUE,
myanCol = mycol) +
AverageHeatmap(object = pbmc,
markerGene = markers$gene,
annoCol = TRUE,
myanCol = mycol1)
markerVocalno is used to plot vocalnoplot of marker genes:
test <- system.file("extdata", "pbmc.markers.csv", package = "scRNAtoolVis")
markers <- read.csv(test)
# plot
markerVocalno(markers = markers,
topn = 5,
labelCol = ggsci::pal_npg()(9))

More parameters refer to:
?clusterCornerAxes
?FeatureCornerAxes
?AverageHeatmap